

Puntos más importantes:Un árbol filogenético es un diagrama que representa las relaciones evolutivas entre organismos. Los árboles filogenéticos son hipótesis, no hechos definitivos.El patrón de ramificación en un árbol filogenético refleja cómo las especies u otros grupos evolucionaron a partir de una serie de ancestros comunes.En los árboles, dos especies están más relacionadas si tienen un ancestro común más reciente y menos relacionadas si tienen un ancestro común menos reciente.Los árboles filogenéticos pueden dibujarse en varios estilos equivalentes. Rotar un árbol alrededor de sus puntos de ramificación no cambia la información que contiene.IntroducciónLos humanos como grupo somos muy buenos para organizar cosas. No necesariamente cosas como armarios o habitaciones, que personalmente confieso me cuestan mucho trabajo. En cambio, a la gente a menudo le gusta agrupar y ordenar las cosas que ve en el mundo que le rodea. Empezando por el filósofo griego Aristóteles, este deseo de clasificar se ha extendido a los muchos y diversos seres vivos de la tierra.Los sistemas de clasificación más modernos se basan en las relaciones evolutivas entre organismos, esto es, en su filogenia. Los sistemas de clasificación basados en la filogenia organizan las especies u otros grupos de manera que reflejen nuestra comprensión de su proceso evolutivo a partir de sus ancestros comunes.En este artículo, daremos un vistazo a los árboles filogenéticos, diagramas que representan las relaciones evolutivas entre organismos. Veremos exactamente qué podemos (y que no podemos) inferir a partir de un árbol filogenético, así como qué significa que los organismos estén más o menos relacionados en el contexto de estos árboles.Anatomía de un árbol filogenéticoCuando dibujamos un árbol filogenético, estamos representando nuestra mejor hipótesis sobre cómo evolucionó un conjunto de especies (u otros grupos) a partir de un ancestro común^11start superscript, 1, end superscript. Como veremos en el artículo sobre , esta hipótesis se basa en la información que hemos recopilado acerca de nuestro conjunto de especies, cosas como sus características físicas y la secuencia de ADN de sus genes.En un árbol filogenético, las especies o grupos de interés se encuentran en los extremos de las líneas a las que consideramos las ramas del árbol. Por ejemplo, el árbol filogenético siguiente representa las relaciones entre cinco especies A, B, C, D y E, las cuales se ubican en las puntas de las ramas: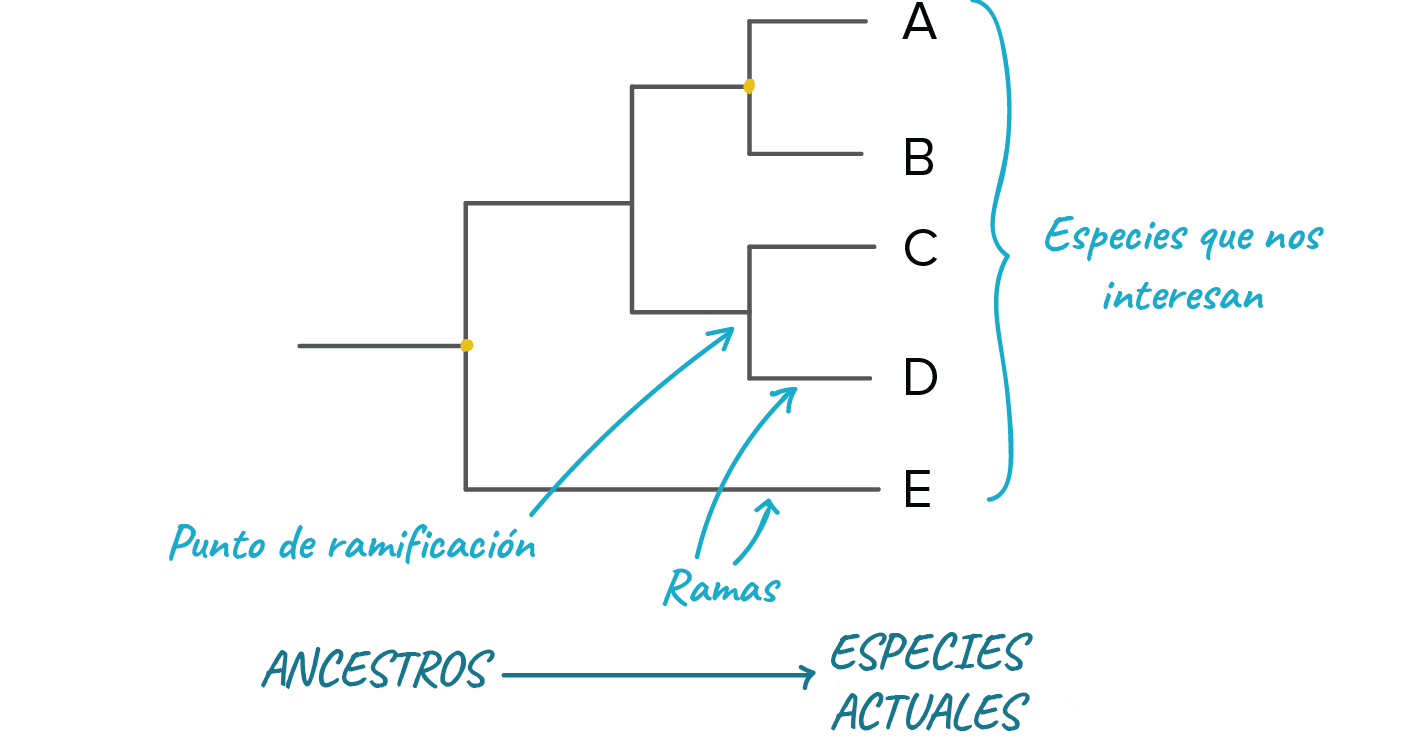 Imagen modificada de de Robert Bear et al., El patrón en el que se conectan ramas representa nuestra comprensión de cómo evolucionaron las especies del árbol a partir de una serie de ancestros comunes. Cada punto de ramificación (también llamado nodo interno) representa un evento de divergencia o separación de un grupo en dos grupos descendientes.En cada punto de ramificación se encuentra el ancestro común más recientede todos los grupos que descienden de esa ramificación. Por ejemplo, en el punto de ramificación que conduce a las especies A y B, encontraríamos al ancestro común más reciente de esas dos especies. En el punto de ramificación que se encuentra justo por arriba de la raíz del árbol, encontraríamos al ancestro común más reciente de todas las especies en el árbol (A, B, C, D, E).
Imagen modificada de de Robert Bear et al., El patrón en el que se conectan ramas representa nuestra comprensión de cómo evolucionaron las especies del árbol a partir de una serie de ancestros comunes. Cada punto de ramificación (también llamado nodo interno) representa un evento de divergencia o separación de un grupo en dos grupos descendientes.En cada punto de ramificación se encuentra el ancestro común más recientede todos los grupos que descienden de esa ramificación. Por ejemplo, en el punto de ramificación que conduce a las especies A y B, encontraríamos al ancestro común más reciente de esas dos especies. En el punto de ramificación que se encuentra justo por arriba de la raíz del árbol, encontraríamos al ancestro común más reciente de todas las especies en el árbol (A, B, C, D, E). 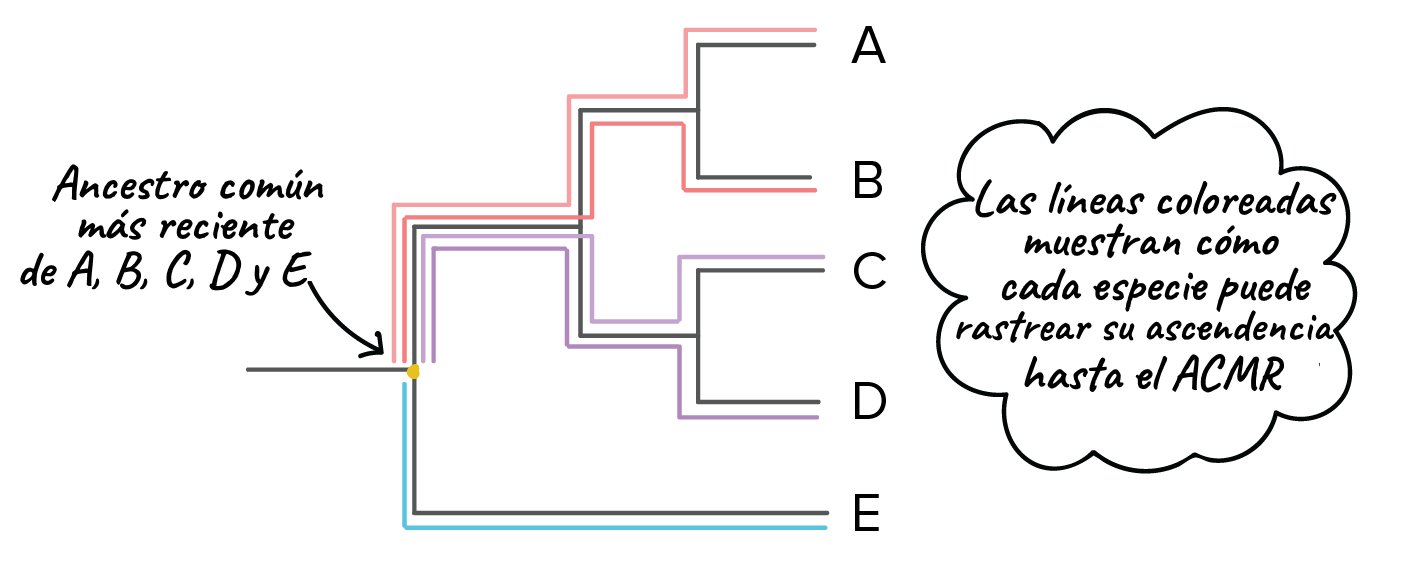
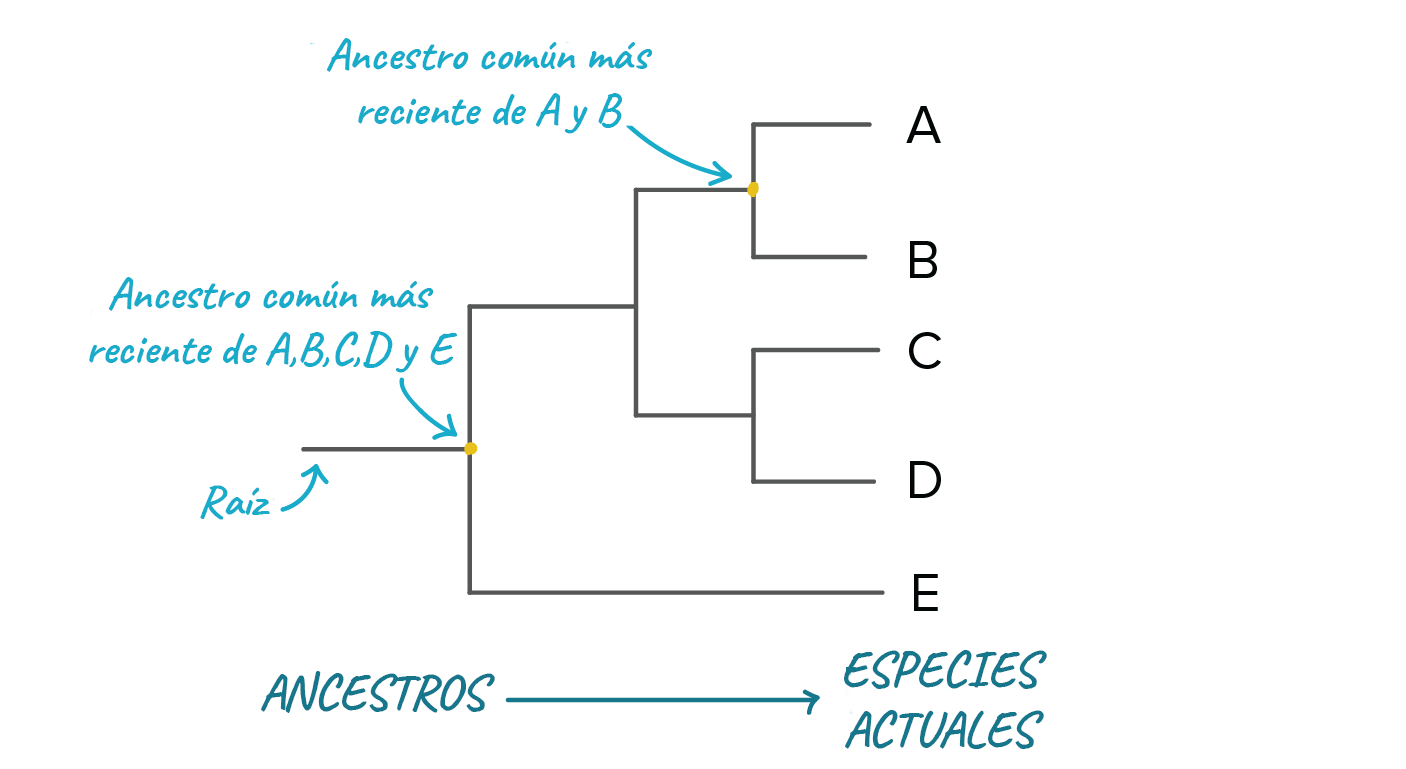 Imagen modificada de de Robert Bear et al., Cada línea horizontal en nuestro árbol representa una serie de ancestros que al final lleva una especie. Por ejemplo, la línea que lleva hacia la especie E representa a los ancestros de la especie desde que divergió de las otras especies en el árbol. De manera similar, la raíz representa una serie de ancestros que conducen hasta el ancestro común más reciente de todas las especies en el árbol.¿Qué especies están más relacionadas?En un árbol filogenético, la relación entre dos especies tiene un significado muy específico. Dos especies están más relacionadas si tienen un ancestro común más reciente y menos relacionadas si tienen un ancestro común menos reciente.Podemos usar un método bastante directo para encontrar al ancestro común más reciente de cualquier par o grupo de especies. En este método, empezamos en la rama en cuyos extremos se encuentran las dos especies de nuestro interés y "retrocedemos" en el árbol hasta que encontramos el punto donde convergen las líneas de ambas especies.Por ejemplo, supón que queremos saber qué especies están más cercanamente relacionadas, si A y B o B y C. Para hacerlo, seguiríamos las líneas de ambos pares de especies hacia atrás en el árbol. Dado que A y B convergen primero en un ancestro común, y que B solo se une con C después de su punto de unión con A, podemos decir que A y B están más relacionadas que B y C.
Imagen modificada de de Robert Bear et al., Cada línea horizontal en nuestro árbol representa una serie de ancestros que al final lleva una especie. Por ejemplo, la línea que lleva hacia la especie E representa a los ancestros de la especie desde que divergió de las otras especies en el árbol. De manera similar, la raíz representa una serie de ancestros que conducen hasta el ancestro común más reciente de todas las especies en el árbol.¿Qué especies están más relacionadas?En un árbol filogenético, la relación entre dos especies tiene un significado muy específico. Dos especies están más relacionadas si tienen un ancestro común más reciente y menos relacionadas si tienen un ancestro común menos reciente.Podemos usar un método bastante directo para encontrar al ancestro común más reciente de cualquier par o grupo de especies. En este método, empezamos en la rama en cuyos extremos se encuentran las dos especies de nuestro interés y "retrocedemos" en el árbol hasta que encontramos el punto donde convergen las líneas de ambas especies.Por ejemplo, supón que queremos saber qué especies están más cercanamente relacionadas, si A y B o B y C. Para hacerlo, seguiríamos las líneas de ambos pares de especies hacia atrás en el árbol. Dado que A y B convergen primero en un ancestro común, y que B solo se une con C después de su punto de unión con A, podemos decir que A y B están más relacionadas que B y C.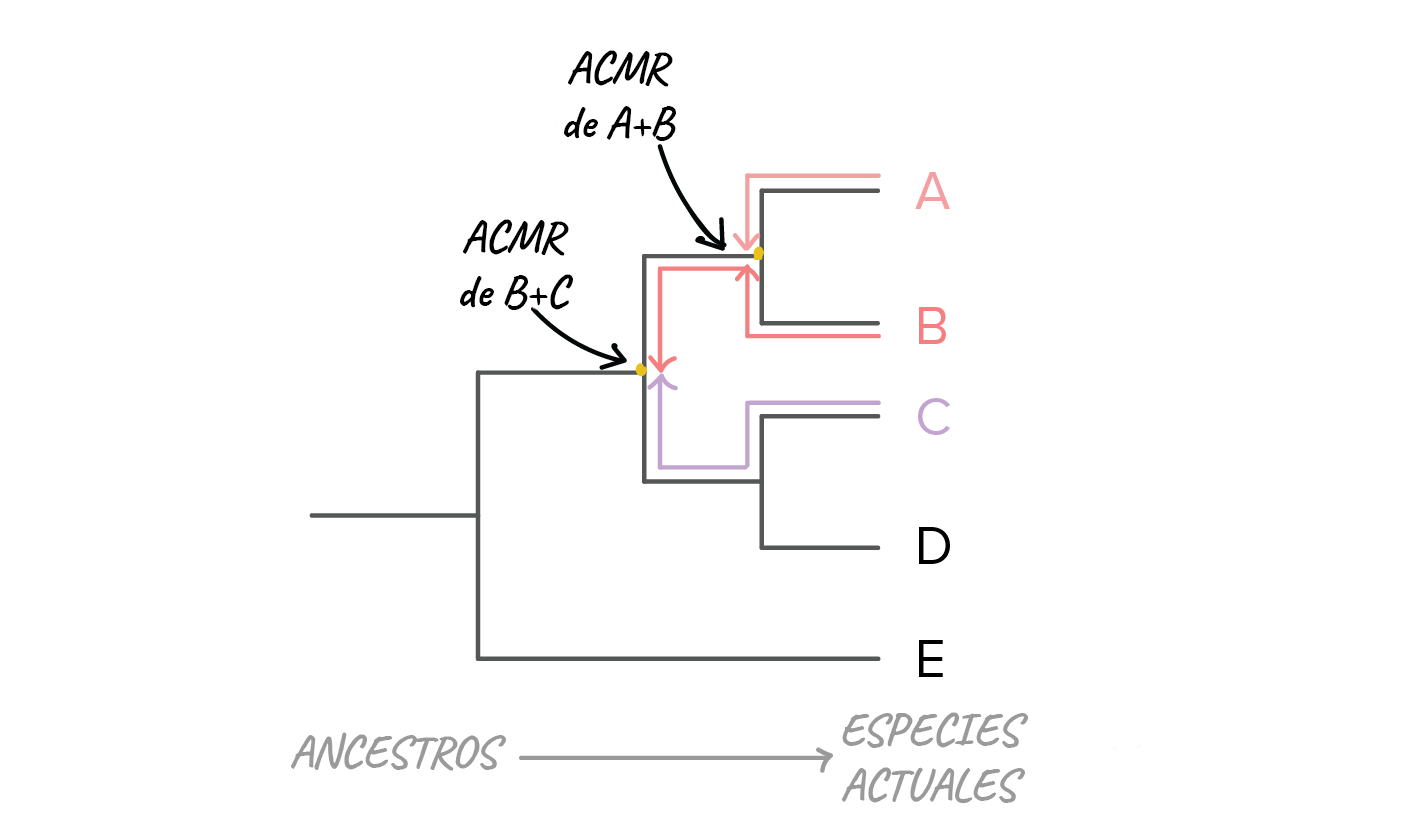 Imagen modificada de de Robert Bear et al., Es importante destacar que hay algunas especies cuyo parentesco no podemos comparar usando este método. Por ejemplo, no podemos decir si A y B están más relacionadas que C y D. Esto es porque, por defecto, el eje horizontal del árbol no representa el tiempo de manera directa. Así que solo podemos comparar el momento de ramificación de los eventos que ocurren en el mismo linaje (misma línea directa desde la raíz del árbol) y no los que suceden en diferentes linajes.Algunas recomendaciones para leer árboles filogenéticosPuedes ver árboles filogenéticos dibujados en muchos formatos diferentes. Algunos usan bloques, como el árbol inferior izquierdo. Otros usan líneas diagonales, como el árbol inferior derecho. También puedes ver árboles de cualquiera de estos tipos orientados de manera vertical o volteados lateralmente, como se muestra el árbol de bloques.
Imagen modificada de de Robert Bear et al., Es importante destacar que hay algunas especies cuyo parentesco no podemos comparar usando este método. Por ejemplo, no podemos decir si A y B están más relacionadas que C y D. Esto es porque, por defecto, el eje horizontal del árbol no representa el tiempo de manera directa. Así que solo podemos comparar el momento de ramificación de los eventos que ocurren en el mismo linaje (misma línea directa desde la raíz del árbol) y no los que suceden en diferentes linajes.Algunas recomendaciones para leer árboles filogenéticosPuedes ver árboles filogenéticos dibujados en muchos formatos diferentes. Algunos usan bloques, como el árbol inferior izquierdo. Otros usan líneas diagonales, como el árbol inferior derecho. También puedes ver árboles de cualquiera de estos tipos orientados de manera vertical o volteados lateralmente, como se muestra el árbol de bloques.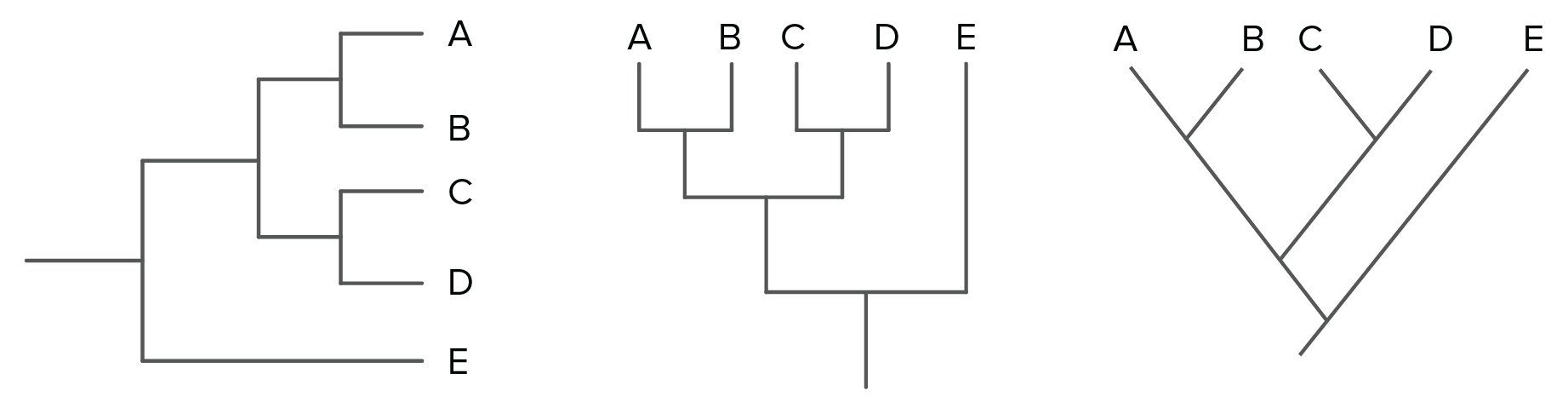 Imagen modificada de de Robert Bear et al., Los dos árboles de arriba representan relaciones idénticas entre las especies A, B, C, D y E. Puede que quieras tomarte un momento para convencerte que así es, que no hay diferencias en el patrón de ramificación ni en lo recientes que son los ancestros comunes en ambos árboles. La información idéntica en estos árboles de apariencia distinta nos recuerda que lo importante en un árbol típico es el patrón de ramificación (y no la longitud de sus ramas).Otro punto crítico acerca de estos árboles es que si rotas las estructuras usando uno de los puntos de ramificación como pivote, no cambias las relaciones. Del mismo modo que los dos árboles de arriba muestran las mismas relaciones aunque su formato sea distinto, todos los árboles de abajo muestran las mismas relaciones entre cuatro especies:
Imagen modificada de de Robert Bear et al., Los dos árboles de arriba representan relaciones idénticas entre las especies A, B, C, D y E. Puede que quieras tomarte un momento para convencerte que así es, que no hay diferencias en el patrón de ramificación ni en lo recientes que son los ancestros comunes en ambos árboles. La información idéntica en estos árboles de apariencia distinta nos recuerda que lo importante en un árbol típico es el patrón de ramificación (y no la longitud de sus ramas).Otro punto crítico acerca de estos árboles es que si rotas las estructuras usando uno de los puntos de ramificación como pivote, no cambias las relaciones. Del mismo modo que los dos árboles de arriba muestran las mismas relaciones aunque su formato sea distinto, todos los árboles de abajo muestran las mismas relaciones entre cuatro especies: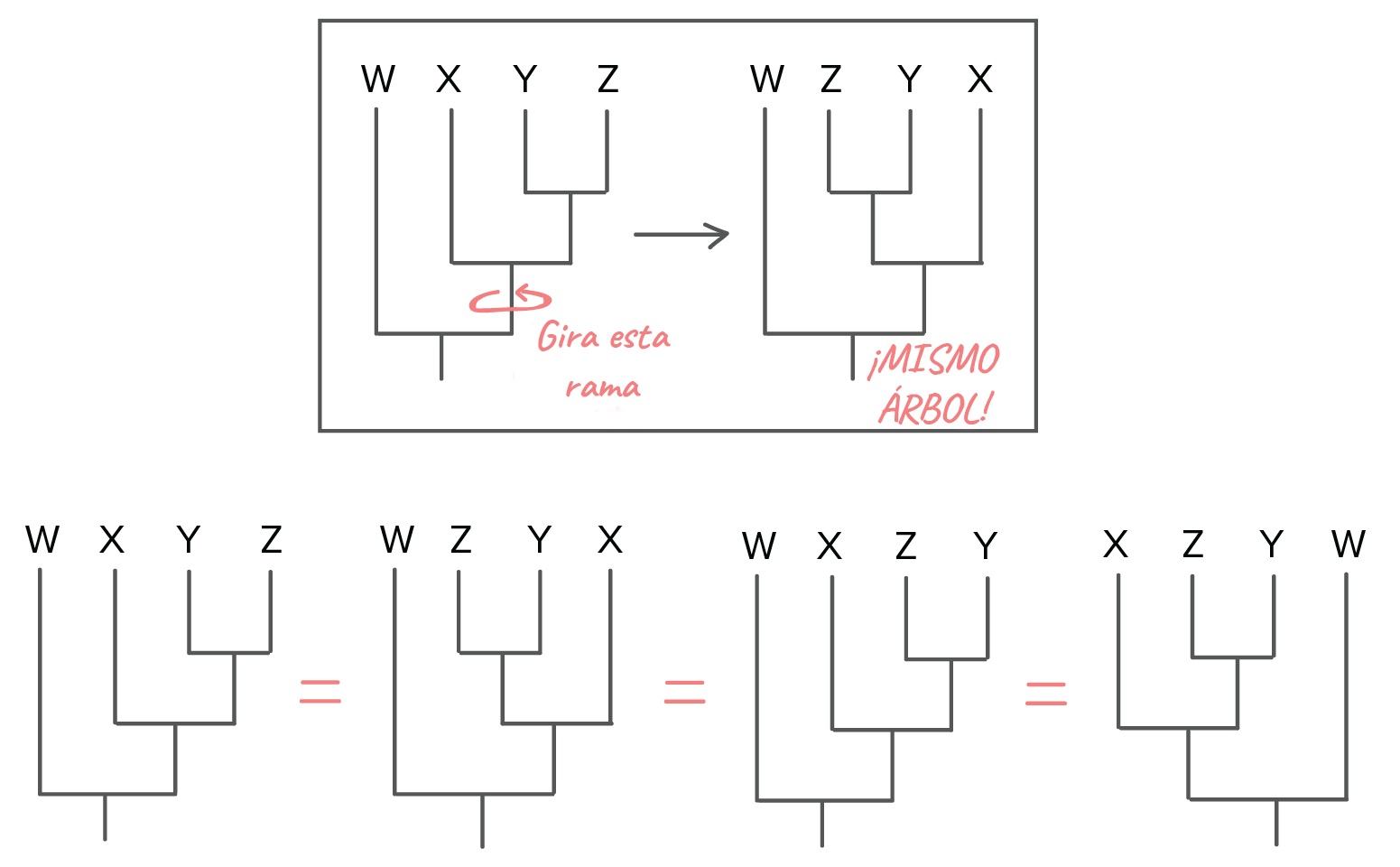 Imagen modificada de de Robert Bear et al., Si no logras ver de inmediato que lo anterior es cierto (¡a primera vista, yo tampoco pude!), solo concéntrate en las relaciones y los puntos de ramificación en lugar del orden de las especies (W, X, Y y Z) en la parte superior de los diagramas. Ese orden en realidad no nos da información útil. En cambio, es la estructura de las ramas de cada diagrama la que nos dice lo que necesitamos para entender el árbol.Hasta ahora, todos los árboles que hemos visto tienen patrones de ramificación limpios y bonitos, con solo dos linajes (líneas de descendencia) que surgen de cada punto de ramificación. Sin embargo, puedes encontrar árboles con una politomía (poli, muchos; tomía, corte), lo que significa que un punto de ramificación tiene tres o más especies diferentes que surgen de él^22start superscript, 2, end superscript. En general, una politomía nos muestra dónde no tenemos suficiente información para determinar el orden de las ramas.
Imagen modificada de de Robert Bear et al., Si no logras ver de inmediato que lo anterior es cierto (¡a primera vista, yo tampoco pude!), solo concéntrate en las relaciones y los puntos de ramificación en lugar del orden de las especies (W, X, Y y Z) en la parte superior de los diagramas. Ese orden en realidad no nos da información útil. En cambio, es la estructura de las ramas de cada diagrama la que nos dice lo que necesitamos para entender el árbol.Hasta ahora, todos los árboles que hemos visto tienen patrones de ramificación limpios y bonitos, con solo dos linajes (líneas de descendencia) que surgen de cada punto de ramificación. Sin embargo, puedes encontrar árboles con una politomía (poli, muchos; tomía, corte), lo que significa que un punto de ramificación tiene tres o más especies diferentes que surgen de él^22start superscript, 2, end superscript. En general, una politomía nos muestra dónde no tenemos suficiente información para determinar el orden de las ramas.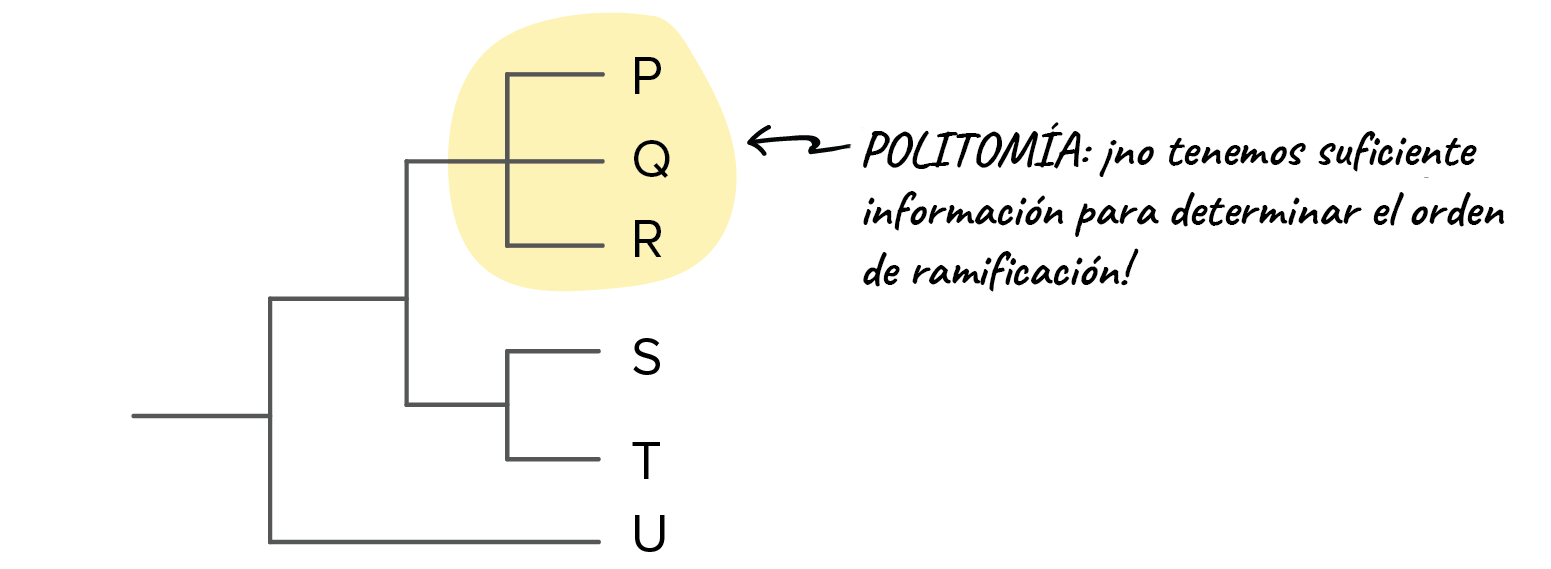 Imagen modificada de de Robert Bear et al., Si más adelante obtenemos más información acerca de las especies en un árbol, es posible que podamos resolver la politomía usando la información nueva.¿De dónde vienen estos árboles?Para generar un árbol filogenético, los científicos a menudo comparan y analizan muchas características de las especies o grupos involucrados. Estas características pueden incluir la morfología externa (forma y apariencia), la anatomía interna, el comportamiento, las rutas bioquímicas, las secuencias de ADN y proteínas, e incluso las características de fósiles.Para construir árboles precisos y significativos, los biólogos a menudo usan muchas características distintas (lo que reduce las posibilidades de que cualquier información imperfecta resulte en un árbol erróneo). Aun así, los árboles filogenéticos son hipótesis, no respuestas definitivas, y son tan buenos como la información disponible con la que fueron construidos. Los árboles se revisan y actualizan con el tiempo a medida que hay nueva información disponible que pueda ser añadida al análisis. Esto es particularmente cierto hoy en día, ya que la secuenciación de ADN aumenta nuestra habilidad de comparar genes entre especies.En el siguiente artículo sobre , veremos ejemplos concretos de cómo se usan los diferentes tipos de información para organizar las especies en árboles filogenéticos.
Imagen modificada de de Robert Bear et al., Si más adelante obtenemos más información acerca de las especies en un árbol, es posible que podamos resolver la politomía usando la información nueva.¿De dónde vienen estos árboles?Para generar un árbol filogenético, los científicos a menudo comparan y analizan muchas características de las especies o grupos involucrados. Estas características pueden incluir la morfología externa (forma y apariencia), la anatomía interna, el comportamiento, las rutas bioquímicas, las secuencias de ADN y proteínas, e incluso las características de fósiles.Para construir árboles precisos y significativos, los biólogos a menudo usan muchas características distintas (lo que reduce las posibilidades de que cualquier información imperfecta resulte en un árbol erróneo). Aun así, los árboles filogenéticos son hipótesis, no respuestas definitivas, y son tan buenos como la información disponible con la que fueron construidos. Los árboles se revisan y actualizan con el tiempo a medida que hay nueva información disponible que pueda ser añadida al análisis. Esto es particularmente cierto hoy en día, ya que la secuenciación de ADN aumenta nuestra habilidad de comparar genes entre especies.En el siguiente artículo sobre , veremos ejemplos concretos de cómo se usan los diferentes tipos de información para organizar las especies en árboles filogenéticos.

